-
Notifications
You must be signed in to change notification settings - Fork 450
Commit
This commit does not belong to any branch on this repository, and may belong to a fork outside of the repository.
- Loading branch information
1 parent
f11c955
commit 2598331
Showing
20 changed files
with
2,263 additions
and
0 deletions.
There are no files selected for viewing
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1,22 @@ | ||
| MIT License | ||
|
|
||
| Copyright (c) 2025 Robotechnic | ||
|
|
||
| Permission is hereby granted, free of charge, to any person obtaining | ||
| a copy of this software and associated documentation files (the | ||
| "Software"), to deal in the Software without restriction, including | ||
| without limitation the rights to use, copy, modify, merge, publish, | ||
| distribute, sublicense, and/or sell copies of the Software, and to | ||
| permit persons to whom the Software is furnished to do so, subject to | ||
| the following conditions: | ||
|
|
||
| The above copyright notice and this permission notice shall be | ||
| included in all copies or substantial portions of the Software. | ||
|
|
||
| THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, | ||
| EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF | ||
| MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND | ||
| NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS BE | ||
| LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION | ||
| OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION | ||
| WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE. |
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1,110 @@ | ||
| # alchemist | ||
|
|
||
| Alchemist is a typst package to draw skeletal formulae. It is based on the [chemfig](https://ctan.org/pkg/chemfig) package. The main goal of alchemist is not to reproduce one-to-one chemfig. Instead, it aims to provide an interface to achieve the same results in Typst. | ||
|
|
||
| <!--EXAMPLE(links)--> | ||
| ````typ | ||
| #skeletize({ | ||
| molecule(name: "A", "A") | ||
| single() | ||
| molecule("B") | ||
| branch({ | ||
| single(angle: 1) | ||
| molecule( | ||
| "W", | ||
| links: ( | ||
| "A": double(stroke: red), | ||
| ), | ||
| ) | ||
| single() | ||
| molecule(name: "X", "X") | ||
| }) | ||
| branch({ | ||
| single(angle: -1) | ||
| molecule("Y") | ||
| single() | ||
| molecule( | ||
| name: "Z", | ||
| "Z", | ||
| links: ( | ||
| "X": single(stroke: black + 3pt), | ||
| ), | ||
| ) | ||
| }) | ||
| single() | ||
| molecule( | ||
| "C", | ||
| links: ( | ||
| "X": cram-filled-left(fill: blue), | ||
| "Z": single(), | ||
| ), | ||
| ) | ||
| }) | ||
| ```` | ||
| 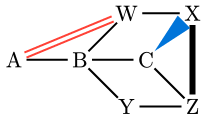 | ||
|
|
||
| Alchemist uses cetz to draw the molecules. This means that you can draw cetz shapes in the same canvas as the molecules. Like this: | ||
|
|
||
| <!--EXAMPLE(cetz)--> | ||
| ````typ | ||
| #skeletize({ | ||
| import cetz.draw: * | ||
| double(absolute: 30deg, name: "l1") | ||
| single(absolute: -30deg, name: "l2") | ||
| molecule("X", name: "X") | ||
| hobby( | ||
| "l1.50%", | ||
| ("l1.start", 0.5, 90deg, "l1.end"), | ||
| "l1.start", | ||
| stroke: (paint: red, dash: "dashed"), | ||
| mark: (end: ">"), | ||
| ) | ||
| hobby( | ||
| (to: "X.north", rel: (0, 1pt)), | ||
| ("l2.end", 0.4, -90deg, "l2.start"), | ||
| "l2.50%", | ||
| mark: (end: ">"), | ||
| ) | ||
| }) | ||
| ```` | ||
| 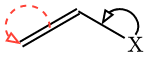 | ||
|
|
||
| ## Usage | ||
|
|
||
| To start using alchemist, just use the following code: | ||
|
|
||
| ```typ | ||
| #import "@preview/alchemist:0.1.4": * | ||
| #skeletize({ | ||
| // Your molecule here | ||
| }) | ||
| ``` | ||
|
|
||
| For more information, check the [manual](https://raw.githubusercontent.com/Robotechnic/alchemist/master/doc/manual.pdf). | ||
|
|
||
| ## Changelog | ||
|
|
||
| ### 0.1.4 | ||
|
|
||
| - Added the possibility to create Lewis formulae | ||
| - Added parenthesis element to create groups and polymer | ||
|
|
||
| ### 0.1.3 | ||
|
|
||
| - Added the possibility to add exponent in the string of a molecule. | ||
|
|
||
| ### 0.1.2 | ||
|
|
||
| - Added default values for link style properties. | ||
| - Updated `cetz` to version 0.3.1. | ||
| - Added a `tip-lenght` argument to dashed cram links. | ||
|
|
||
| ### 0.1.1 | ||
|
|
||
| - Exposed the `draw-skeleton` function. This allows to draw in a cetz canvas directly. | ||
| - Fixed multiples bugs that causes overdraws of links. | ||
|
|
||
| ### 0.1.0 | ||
|
|
||
| - Initial release |
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1,206 @@ | ||
| #import "@preview/cetz:0.3.1" | ||
| #import "src/default.typ": default | ||
| #import "src/utils/utils.typ" | ||
| #import "src/drawer.typ" | ||
| #import "src/drawer.typ": skeletize, draw-skeleton | ||
| #import "src/elements/links.typ": * | ||
| #import "src/elements/molecule.typ": * | ||
| #import "src/elements/lewis.typ": * | ||
|
|
||
| #let transparent = color.rgb(100%, 0, 0, 0) | ||
|
|
||
| /// === Molecule function | ||
| /// Build a molecule group based on mol | ||
| /// Each molecule is represented as an optional count followed by a molecule name | ||
| /// starting by a capital letter followed by an optional exponent followed by an optional indice. | ||
| /// #example(``` | ||
| /// #skeletize({ | ||
| /// molecule("H_2O") | ||
| /// }) | ||
| ///```) | ||
| /// #example(``` | ||
| /// #skeletize({ | ||
| /// molecule("H^A_EF^5_4") | ||
| /// }) | ||
| /// ```) | ||
| /// It is possible to use an equation as a molecule. In this case, the splitting of the equation uses the same rules as in the string case. However, you get some advantages over the string version: | ||
| /// - You can use parenthesis to group elements together. | ||
| /// - You have no restriction about what you can put in exponent or indice. | ||
| /// #example(``` | ||
| /// #skeletize({ | ||
| /// molecule($C(C H_3)_3$) | ||
| /// }) | ||
| ///```) | ||
| /// - name (content): The name of the molecule. It is used as the cetz name of the molecule and to link other molecules to it. | ||
| /// - links (dictionary): The links between this molecule and previous molecules or hooks. The key is the name of the molecule or hook and the value is the link function. See @links. | ||
| /// | ||
| /// Note that the atom-sep and angle arguments are ignored | ||
| /// - lewis (list): The list of lewis structures to draw around the molecules. See @lewis | ||
| /// - mol (string, equation): The string representing the molecule or an equation of the molecule | ||
| /// - vertical (boolean): If true, the molecule is drawn vertically | ||
| /// #example(``` | ||
| /// #skeletize({ | ||
| /// molecule("ABCD", vertical: true) | ||
| /// }) | ||
| ///```) | ||
| /// -> drawable | ||
| #let molecule(name: none, links: (:), lewis: (), vertical: false, mol) = { | ||
| let atoms = if type(mol) == str { | ||
| split-string(mol) | ||
| } else if mol.func() == math.equation { | ||
| split-equation(mol, equation: true) | ||
| } else { | ||
| panic("Invalid molecule content") | ||
| } | ||
|
|
||
| if type(lewis) != array { | ||
| panic("Lewis formulae elements must be in a list") | ||
| } | ||
|
|
||
| ( | ||
| ( | ||
| type: "molecule", | ||
| name: name, | ||
| atoms: atoms, | ||
| links: links, | ||
| lewis: lewis, | ||
| vertical: vertical, | ||
| count: atoms.len(), | ||
| ), | ||
| ) | ||
| } | ||
|
|
||
| /// === Hooks | ||
| /// Create a hook in the molecule. It allows to connect links to the place where the hook is. | ||
| /// Hooks are placed at the end of links or at the beginning of the molecule. | ||
| /// - name (string): The name of the hook | ||
| /// -> drawable | ||
| #let hook(name) = { | ||
| ( | ||
| ( | ||
| type: "hook", | ||
| name: name, | ||
| ), | ||
| ) | ||
| } | ||
|
|
||
| /// === Branch and cycles | ||
| /// Create a branch from the current molecule, the first element | ||
| /// of the branch has to be a link. | ||
| /// | ||
| /// You can specify an angle argument like for links. This angle will be then | ||
| /// used as the `base-angle` for the branch. | ||
| /// | ||
| /// #example(``` | ||
| /// #skeletize({ | ||
| /// molecule("A") | ||
| /// branch({ | ||
| /// single(angle:1) | ||
| /// molecule("B") | ||
| /// }) | ||
| /// branch({ | ||
| /// double(angle: -1) | ||
| /// molecule("D") | ||
| /// }) | ||
| /// single() | ||
| /// double() | ||
| /// single() | ||
| /// molecule("C") | ||
| /// }) | ||
| ///```) | ||
| /// - body (drawable): the body of the branch. It must start with a link. | ||
| /// -> drawable | ||
| #let branch(body, ..args) = { | ||
| if args.pos().len() != 0 { | ||
| panic("Branch takes one positional argument: the body of the branch") | ||
| } | ||
| ((type: "branch", body: body, args: args.named()),) | ||
| } | ||
|
|
||
| /// Create a regular cycle of molecules | ||
| /// You can specify an angle argument like for links. This angle will be then | ||
| /// the angle of the first link of the cycle. | ||
| /// | ||
| /// The argument `align` can be used to force align the cycle according to the | ||
| /// relative angle of the previous link. | ||
| /// | ||
| /// #example(``` | ||
| /// #skeletize({ | ||
| /// cycle(5, { | ||
| /// single() | ||
| /// double() | ||
| /// single() | ||
| /// double() | ||
| /// single() | ||
| /// }) | ||
| /// }) | ||
| ///```) | ||
| /// - faces (int): the number of faces of the cycle | ||
| /// - body (drawable): the body of the cycle. It must start and end with a molecule or a link. | ||
| /// -> drawable | ||
| #let cycle(faces, body, ..args) = { | ||
| if args.pos().len() != 0 { | ||
| panic("Cycle takes two positional arguments: number of faces and body") | ||
| } | ||
| if faces < 3 { | ||
| panic("A cycle must have at least 3 faces") | ||
| } | ||
| ( | ||
| ( | ||
| type: "cycle", | ||
| faces: faces, | ||
| body: body, | ||
| args: args.named(), | ||
| ), | ||
| ) | ||
| } | ||
|
|
||
| /// === Parenthesis | ||
| /// Encapsulate a drawable between two parenthesis. The left parenthesis is placed at the left of the first element of the body and by default the right parenthesis is placed at the right of the last element of the body. | ||
| /// | ||
| /// #example(``` | ||
| /// #skeletize( | ||
| /// config: ( | ||
| /// angle-increment: 30deg | ||
| /// ), { | ||
| /// parenthesis( | ||
| /// l:"[", r:"]", | ||
| /// br: $n$, { | ||
| /// single(angle: 1) | ||
| /// single(angle: -1) | ||
| /// single(angle: 1) | ||
| /// }) | ||
| /// }) | ||
| /// ```) | ||
| /// For more examples, see @examples | ||
| /// | ||
| /// - body (drawable): the body of the parenthesis. It must start and end with a molecule or a link. | ||
| /// - l (string): the left parenthesis | ||
| /// - r (string): the right parenthesis | ||
| /// - align (true): if true, the parenthesis will have the same y position. They will also get sized and aligned according to the body height. If false, they are not aligned and the height argument must be specified. | ||
| /// | ||
| /// - height (float, length): the height of the parenthesis. If align is true, this argument is optional. | ||
| /// - yoffset (float, length, list): the vertical offset of parenthesis. You can also provide a tuple for left and right parenthesis | ||
| /// - xoffset (float, length, list): the horizontal offset of parenthesis. You can also provide a tuple for left and right parenthesis | ||
| /// - right (string): Sometime, it is not convenient to place the right parenthesis at the end of the body. In this case, you can specify the name of the molecule or link where the right parenthesis should be placed. It is especially useful when the body end by a cycle. See @polySulfide | ||
| /// - tr (content): the exponent content of the right parenthesis | ||
| /// - br (content): the indice content of the right parenthesis | ||
| /// -> drawable | ||
| #let parenthesis(body, l: "(", r: ")", align: true, height: none, yoffset: none, xoffset: none, right: none, tr: none, br: none) = { | ||
| ( | ||
| ( | ||
| type: "parenthesis", | ||
| body: body, | ||
| calc: calc, | ||
| l: l, | ||
| r: r, | ||
| align: align, | ||
| tr: tr, | ||
| br: br, | ||
| height: height, | ||
| xoffset: xoffset, | ||
| yoffset: yoffset, | ||
| right: right, | ||
| ), | ||
| ) | ||
| } |
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1,54 @@ | ||
| #let default = ( | ||
| atom-sep: 3em, | ||
| molecule-margin: 0.2em, | ||
| angle-increment: 45deg, | ||
| base-angle: 0deg, | ||
| debug: false, | ||
| single: ( | ||
| stroke: black | ||
| ), | ||
| double: ( | ||
| gap: .25em, | ||
| offset: "center", | ||
| offset-coeff: 0.85, | ||
| stroke: black | ||
| ), | ||
| triple: ( | ||
| gap: .25em, | ||
| stroke: black | ||
| ), | ||
| filled-cram: ( | ||
| stroke: none, | ||
| fill: black, | ||
| base-length: .8em | ||
| ), | ||
| dashed-cram: ( | ||
| stroke: black + .05em, | ||
| dash-gap: .3em, | ||
| base-length: .8em, | ||
| tip-length: .1em | ||
| ), | ||
| lewis-single: ( | ||
| stroke: black, | ||
| fill: black, | ||
| radius: .1em, | ||
| gap: .25em, | ||
| offset: "top" | ||
| ), | ||
| lewis-double: ( | ||
| stroke: black, | ||
| fill: black, | ||
| radius: .1em, | ||
| gap: .25em, | ||
| ), | ||
| lewis-line: ( | ||
| stroke: black, | ||
| length: .7em, | ||
| ), | ||
| lewis-rectangle: ( | ||
| stroke: .08em + black, | ||
| fill: white, | ||
| height: .7em, | ||
| width: .3em | ||
| ) | ||
| ) |
Oops, something went wrong.